BiQ Analyzer Frequently asked questions
Contents
BiQ Analyzer HiMod FAQ
BiQ Analyzer "Classic" FAQ
BiQ Analyzer HiMod FAQ
BiQ Analyzer HiMod doesn't work as expected? There are some hints on how to solve typical problems
Please click on a question in order to expand the answer.
Q: How do I preprocess my data to use it in BiQ Analyzer HiMod?
A: Data files returned from a sequencing machine cannot be used with BiQ Analyzer HiMod directly and have to be preprocessed. To prepare the data we recommend a selection of tools included in Galaxy and provide a step by step guideline on how to use them.
Galaxy can be used in various ways. We recommend using the main public Galaxy server which can be accessed at https://usegalaxy.org/. Other options to use Galaxy are a local installation, a cloud instance, using it on Slipstream or using one of the many other server based versions. All these options can be chosen at http://galaxyproject.org/. In this tutorial we describe the usage of the main public server option.
After opening Galaxy, one can see a list of tools on the left side and a history of loaded and processed files on the right. The space in the middle shows the currently open file or result.
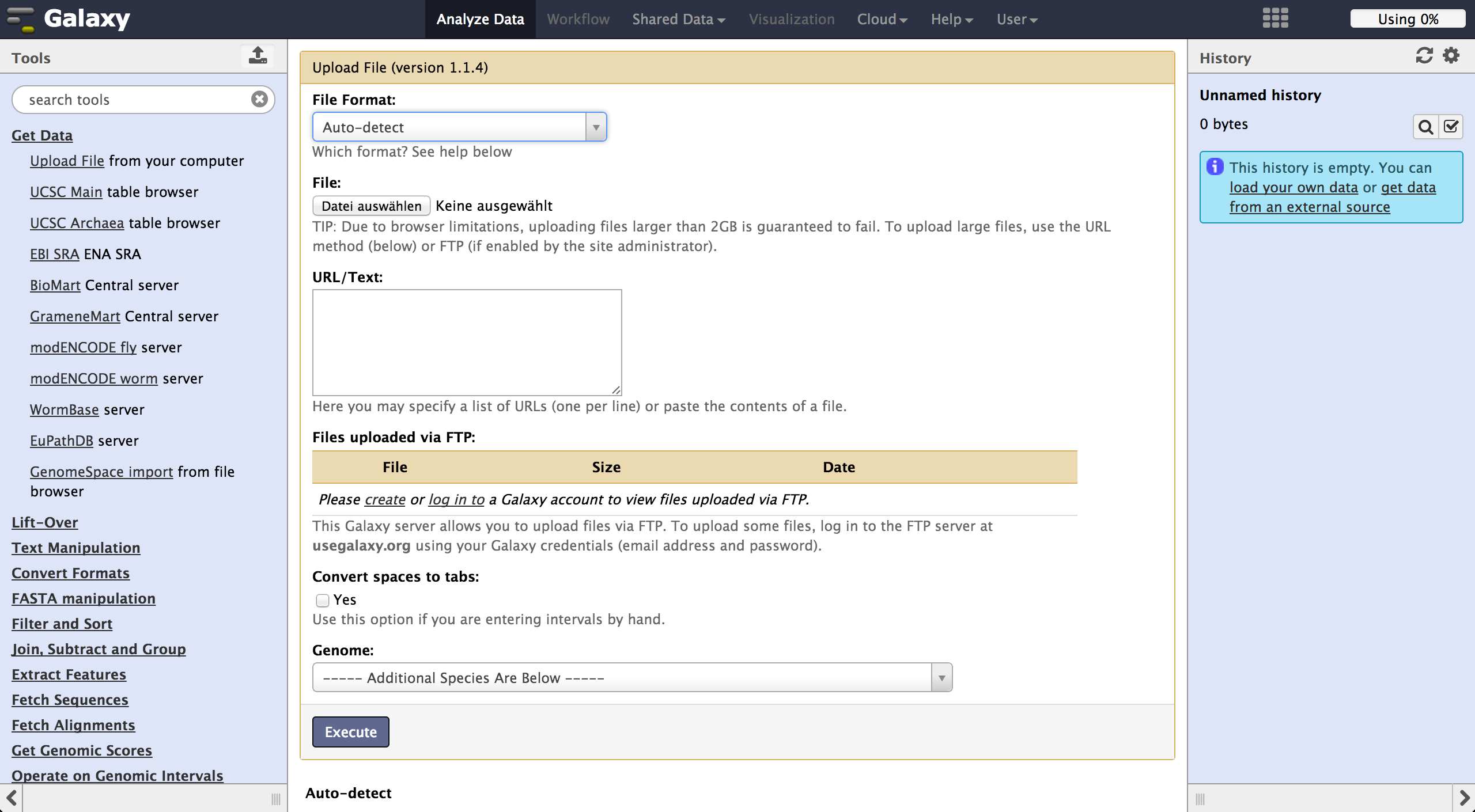
To load files the user can choose "Get Data" -> "Upload File". The file format should be automatically detected but the user can specify it if necessary. Files can be uploaded from local harddisc, by URL or via a FTP Server. The user can further specify the genome of their reads and let the tool automatically convert spaces to tabs while uploading the file. Uploaded files appear consecutively numbered in the history on the right side and can be accessed there.
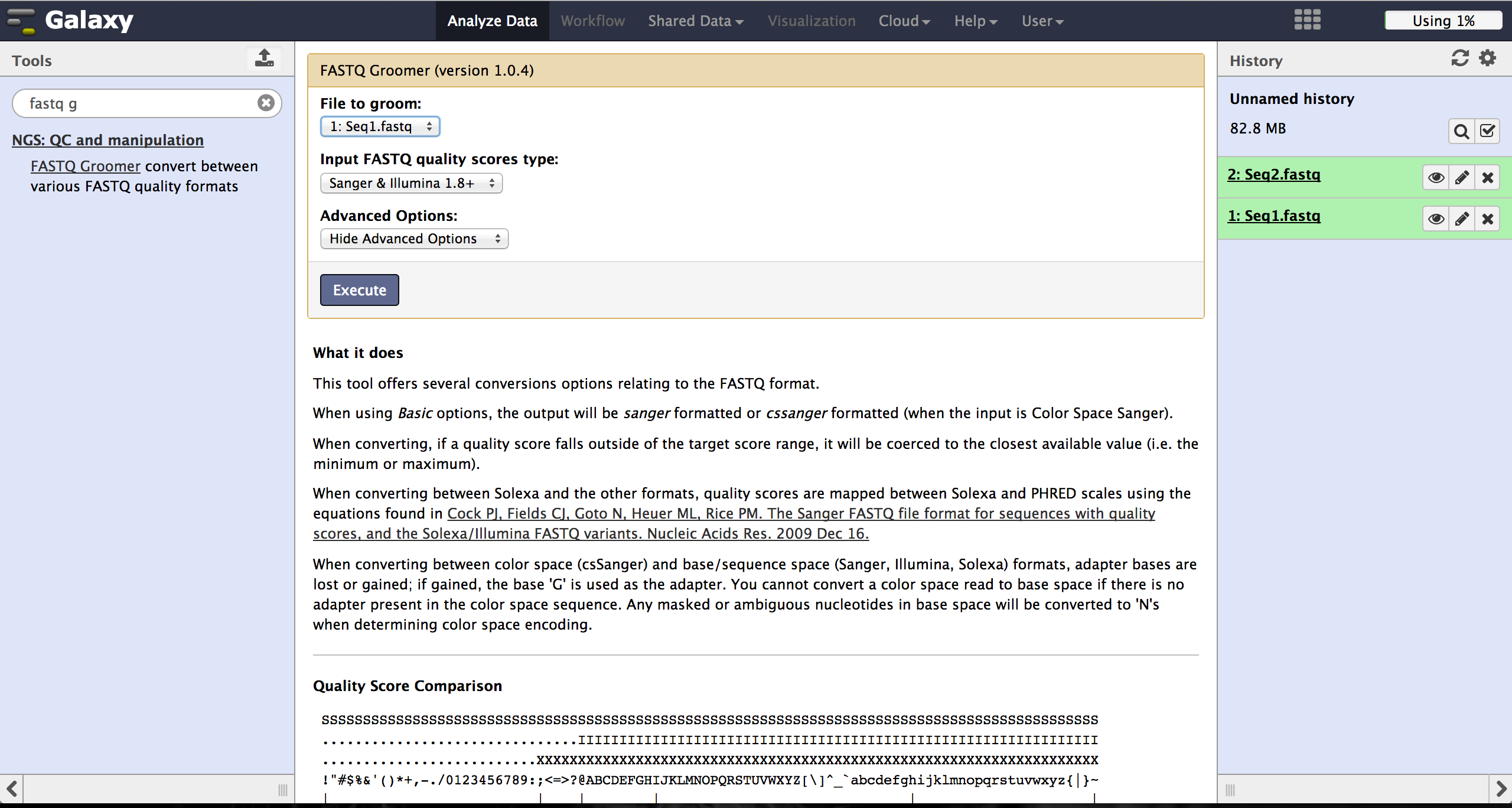
FastQ quality encoding - FASTQ Groomer
BiQ Analyzer HiMod's quality filter supports only Sanger or Illumina 1.8+ encoding. The FASTQ Groomer can be used to convert encodings. This tool can be found at "NGS: QC and manipulation" -> "FASTQ Groomer". Setting a loaded file and a given encoding style, the FASTQ Groomer will change the encoding to Sanger, if nothing else is specified in the advanced options. If the loaded file is already in Sanger format, it will not be changed. After using the FASTQ Groomer on a file, a new file with a name containing "[]FASTQ Groomer on data[]" will appear in the history. In case of working with paired end sequencing data, it is necessary to use this tool as a first step. The clipping tool is very sensitive about quality encoding and will only work on Sanger files.
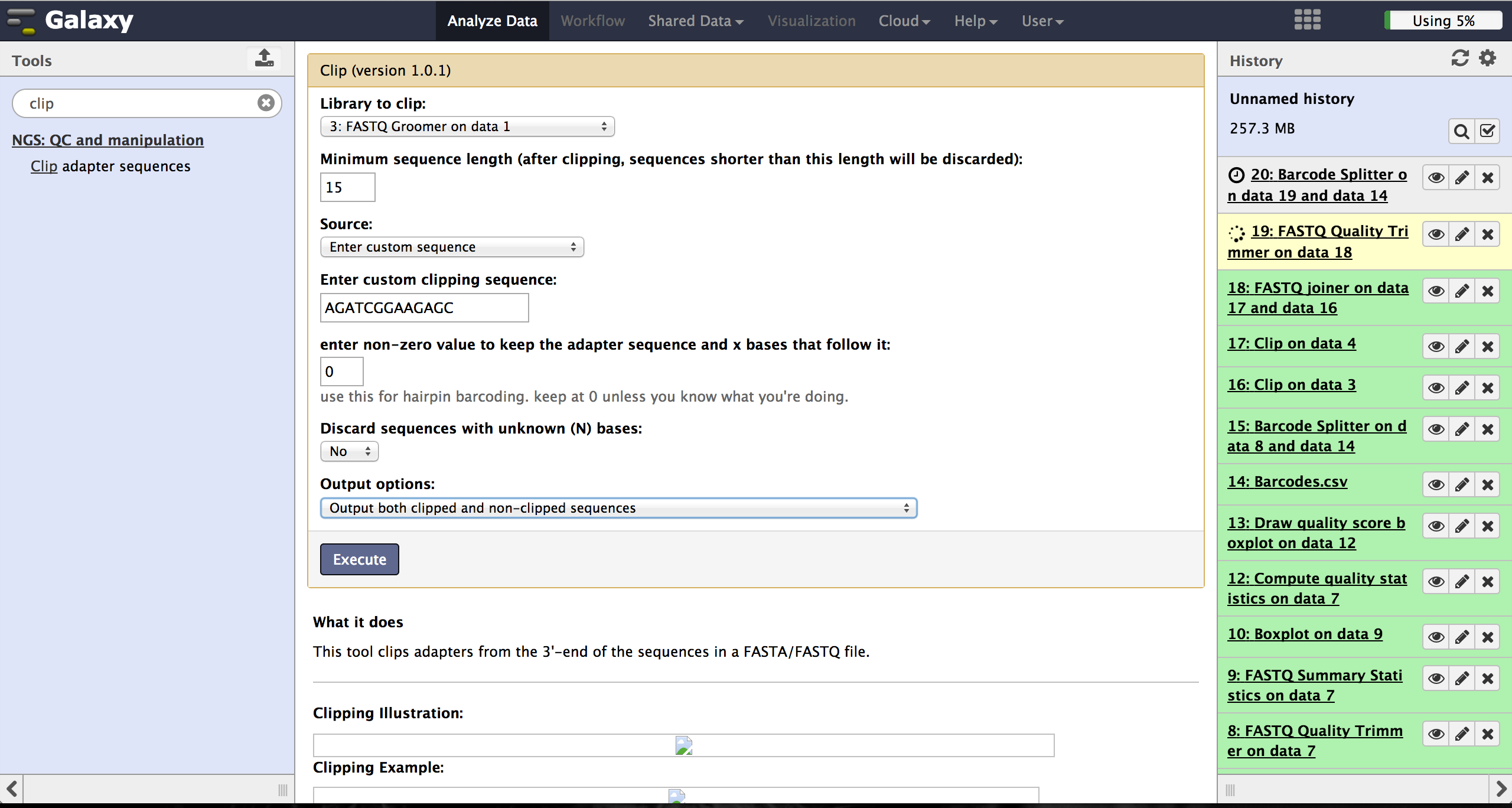
Clipping Adapter sequences - Clip
In case of a sequencing approach with paired ends, the user will end up with two files per lane. One contains the reads started at the 5'end and the other the reads started at the 3' end. The Clip tool - accessible at "NGS: QC and manipulation" -> "Clip" - removes adapter sequences to prepare the joining step. The user has to enter a groomed file and choose an adapter sequence. The standard Illumina adapter sequence is "AGATCGGAAGAGC". It is further necessary to specify which reads should be kept and which should be discarded based on the clipping results.
Joining paired reads - FASTQ joiner
The FASTQ joiner - accessible at "NGS: QC and manipulation" -> "FASTQ joiner" - can join two files of a paired end sequencing and produce one file containing single end reads. Therefore the user has to specify the two matching files for each lane and run the tool.
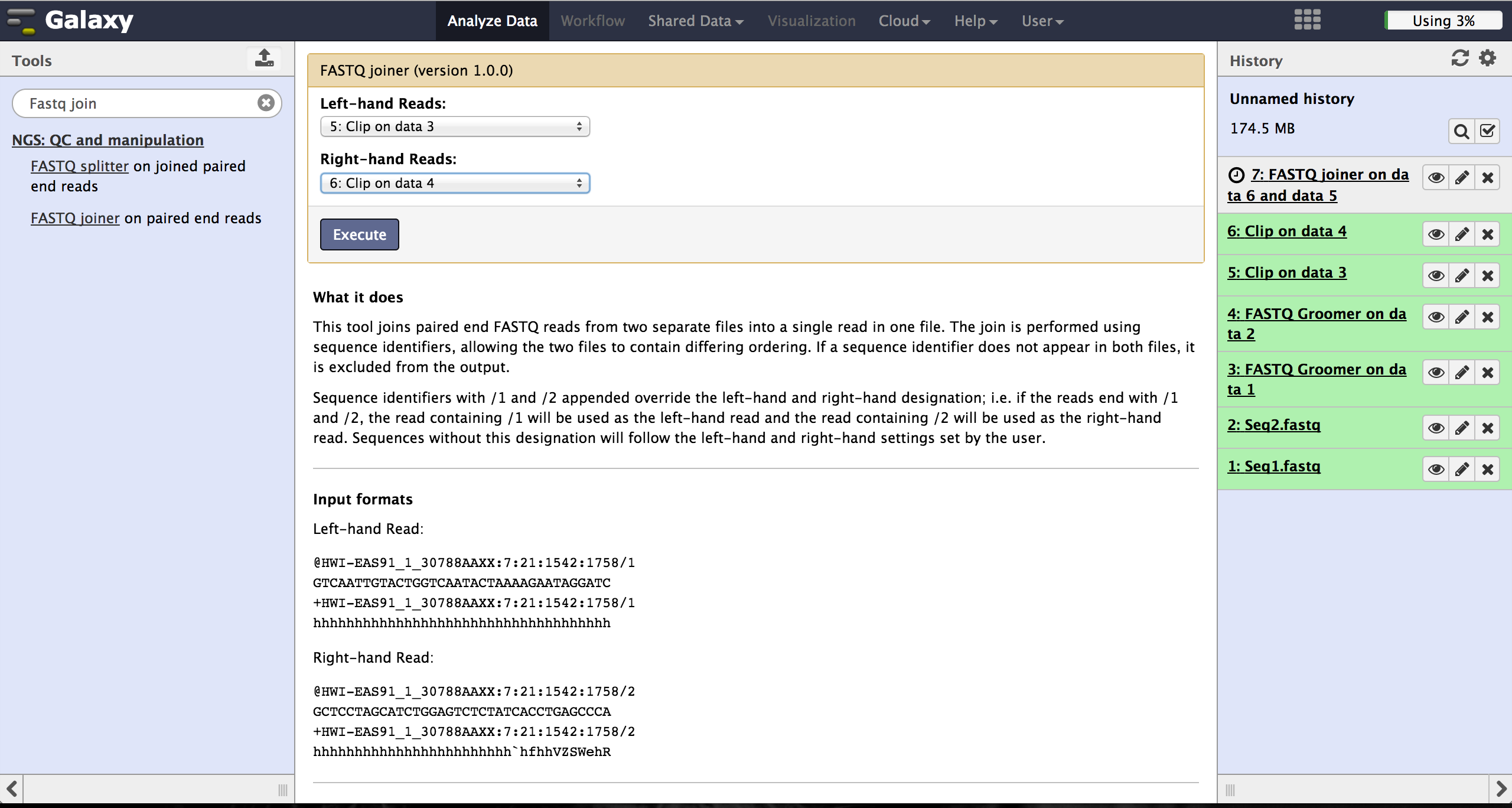
Trimming bad quality bases at read ends - FASTQ Quality Trimmer
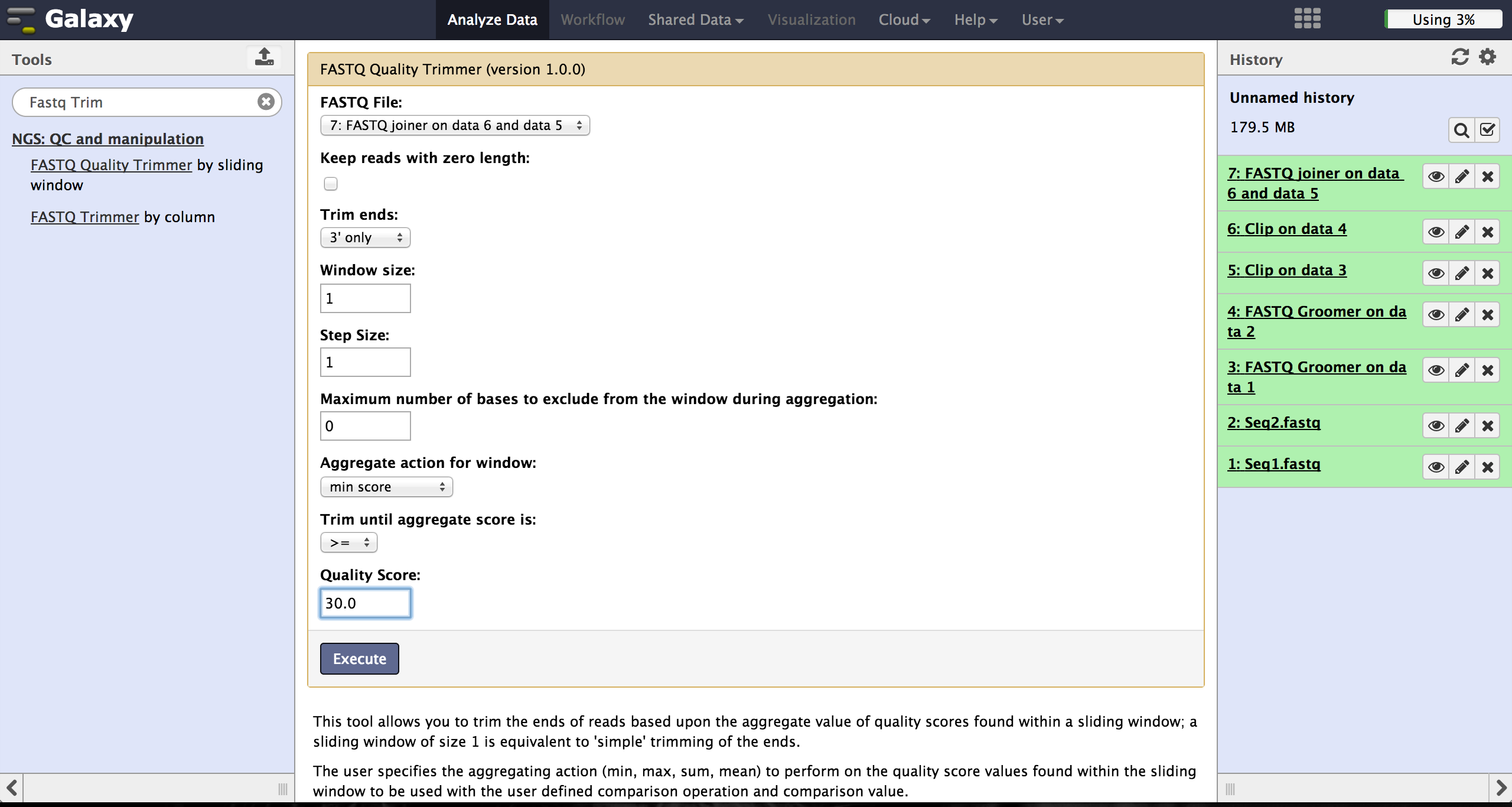
The user can choose to trim reads that have ends with bad quality scores. The FASTQ Quality Trimmer tool ("NGS: QC and manipulation" -> "FASTQ Quality Trimmer") uses a sliding window approach to check for bases with bad quality and trims them until he reaches an area with a good quality. The user has to specify the current FastQ file and a minimal quality score that should be reached. Window size, step size and other parameters can also be set, but the default parameters lead to usable results. One should only trim from the 3' end, because the 5' end contains the primer sequences needed to demultiplex later on and should therefore not be altered. The 5' end can be trimmed in the end after demultiplexing.
Trimming bad quality bases at read ends - FASTQ Trimmer
The FASTQ Trimmer ("NGS: QC and manipulation" -> "FASTQ Trimmer") deletes a specified
number of bases from each end of every read. This is another more strict method to get rid
of bad quality bases but comes with a loss of good bases at the same position. We
recommend the method using the FASTQ Quality Trimmer tool described above.
To find out a good parameter for each end, the user has to use FASTQ Summary
Statistics first. This can be found at "NGS: QC and manipulation" -> "FASTQ joiner".
Passing the joined FastQ file to this toll and executing it results in a table file
that can be used to produce a boxplot using Boxplot. Boxplot can be found at
"Graph/Display Data" -> "Boxplot". The user has to choose the Summary Statistics file and
start the tool. All other parameters can be left untouched.
By manually inspecting the boxplot, the user can decide which bases need to be
trimmed to reach a good enough quality score.
The results for 5'end and 3'end can be passed to the FASTQ Trimmer as well as the
current FastQ file. The run of the tool will result in a new trimmed FastQ file.
In this case the user should also only trim from the 3' end to not alter the primer sequence
on the 5' end.
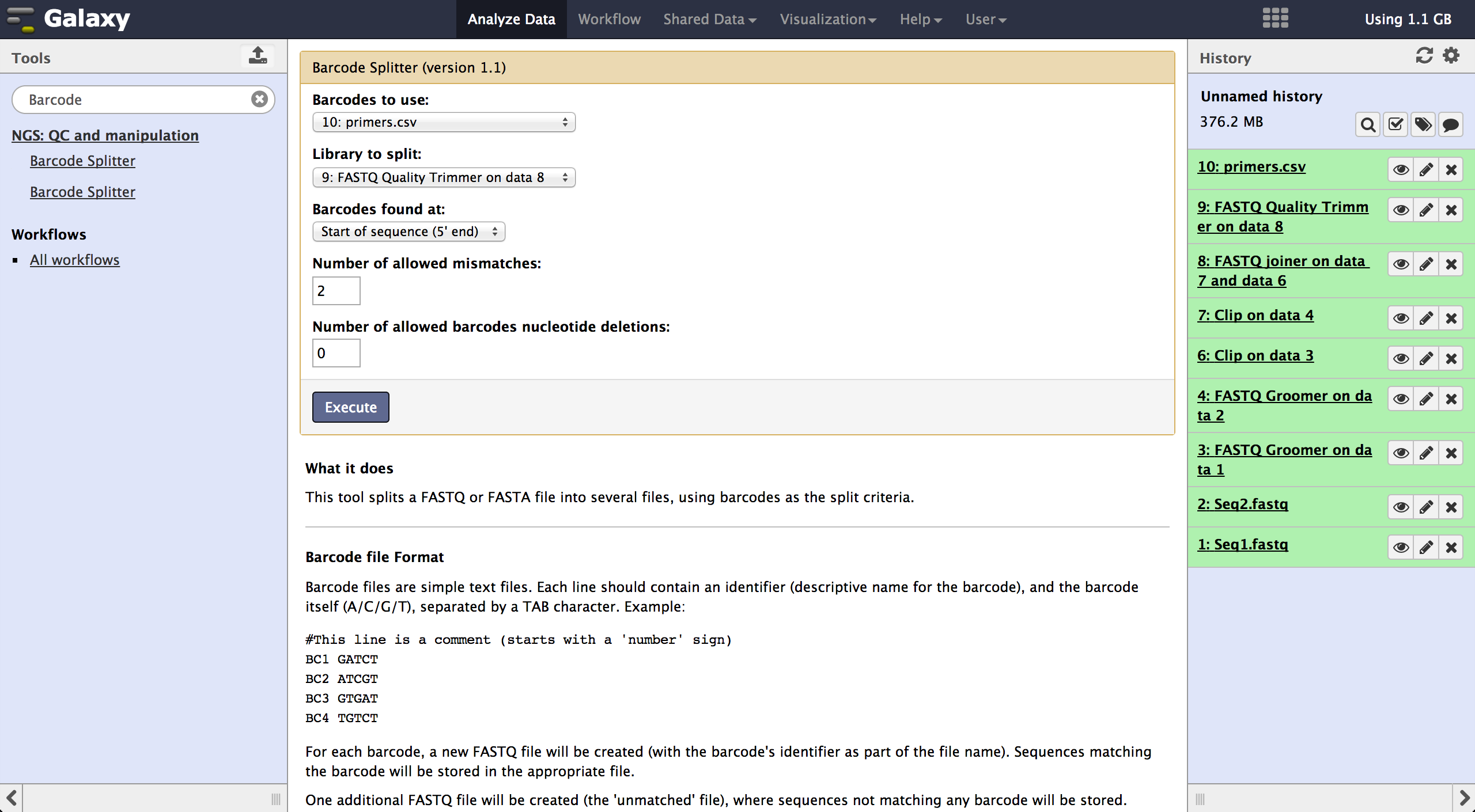
Demultiplexing - Barcode Splitter Version 1.0.0
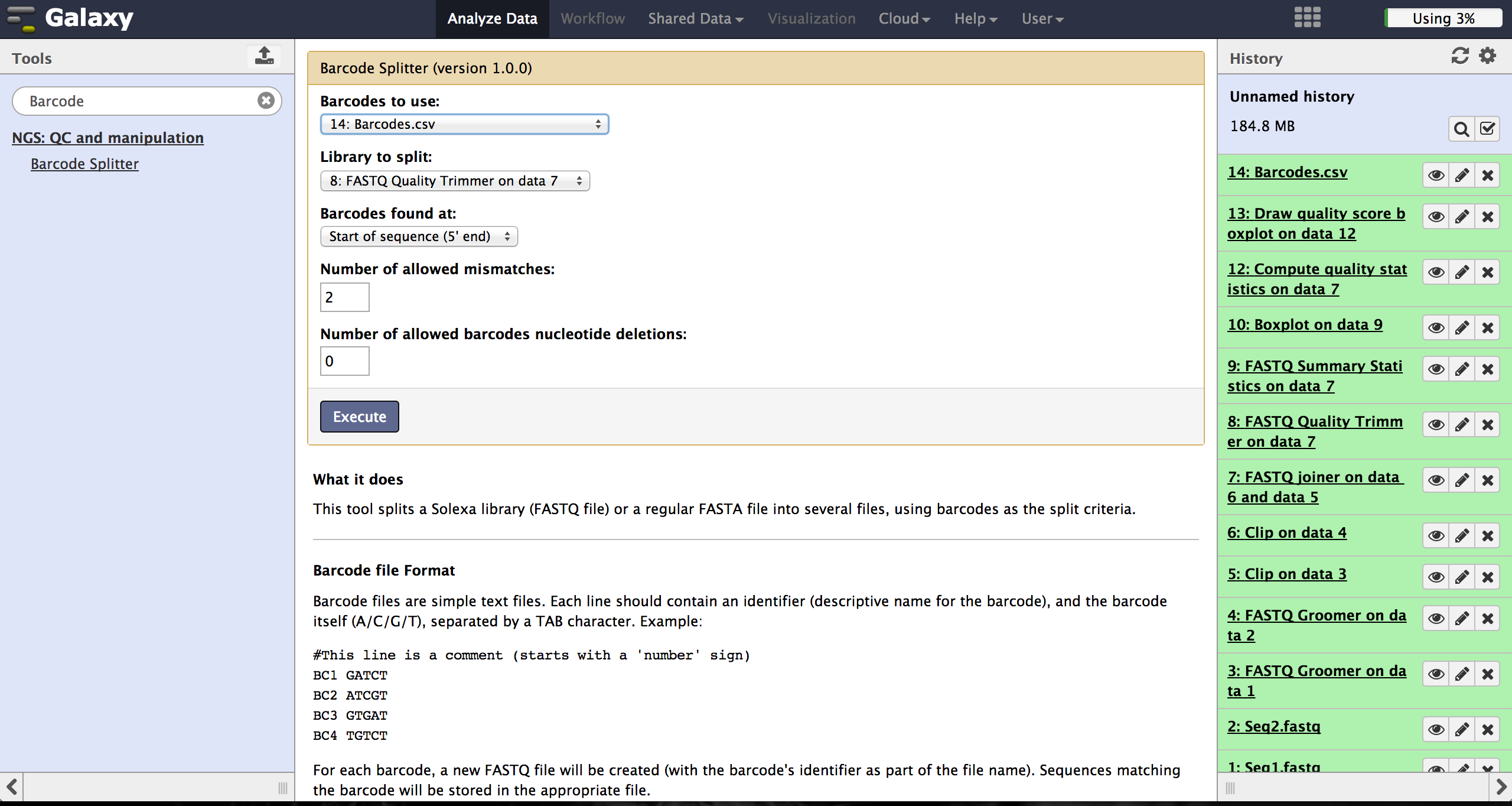
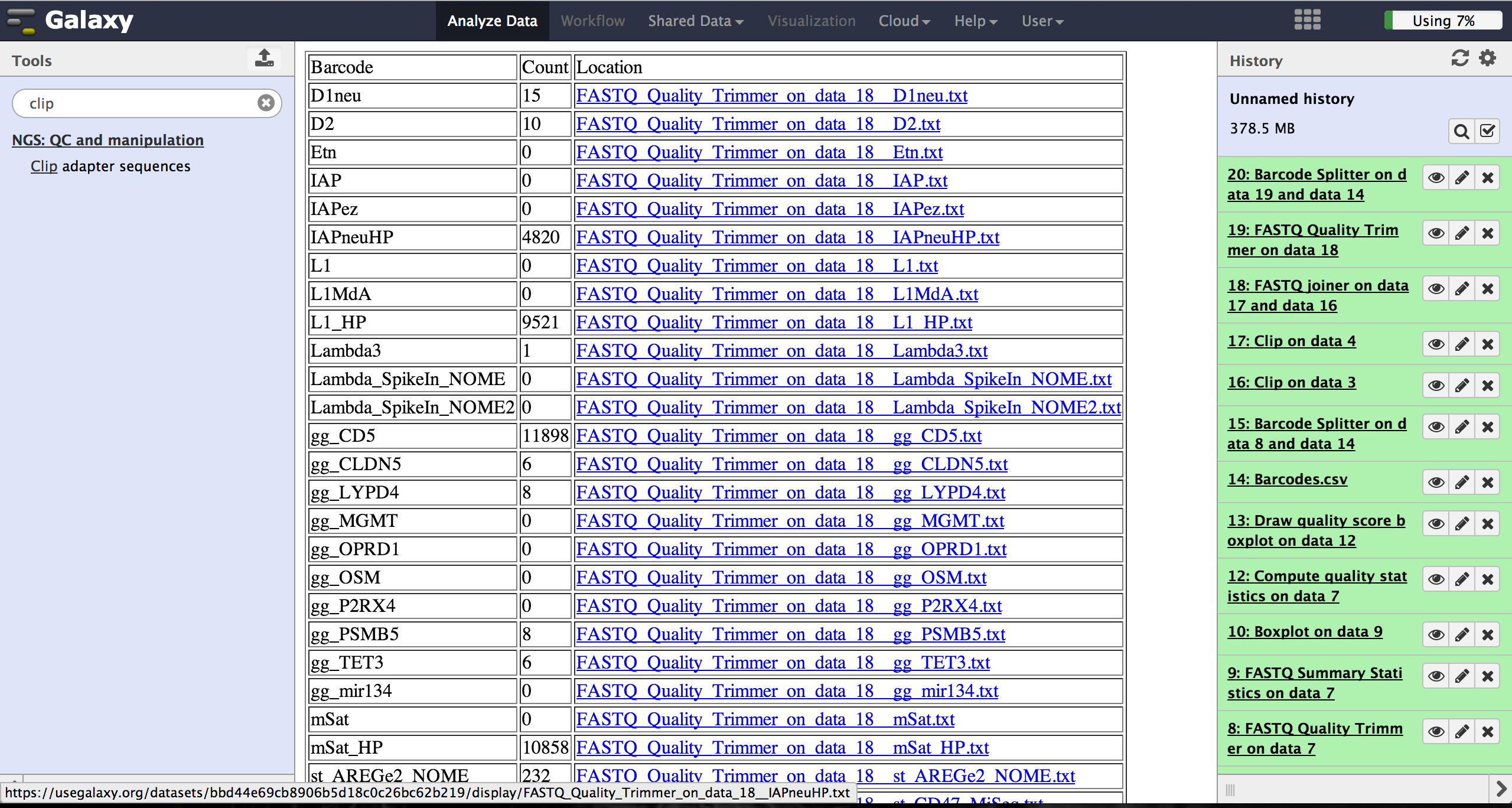
As a last step it is necessary to demultiplex the FastQ file, if more than one amplicon was sequenced per lane. To do so the user has to upload another file containing the primers - which act as barcodes in this case - belonging to the different amplicons. Such a file should be tab separated and contain in each line an identifier for the amplicon and the primer. All primers have to have the same length. The Barcode Splitter 1.0.0 can be found at "NGS: QC and manipulation" -> "Barcode Splitter". Specifying the barcode file, the current FastQ file and at which end of the reads the primers can be found, the user can start the demultiplexing. The number of allowed mismatches and the number of allowed primer nucleotide deletions can be given to improve the results. A new file appears in the history containing a table of the different identifiers, how many reads matched this identifier and a link to a file containing these reads. Opening such a file and clicking the download button on the demultiplexing result file in the history downloads a zip archive including all new FastQ files. The filename extension of those is txt but they contain FastQ data and can therefore be easily renamed. Sequencing platforms as Illumina MiSeq and HiSeq demultiplex by barcodes itself resulting in FastQ files, each containing only reads for one barcode per amplicon. The barcode is given in the first line of each read. In case the raw read files come from a platform which does not do this automatically, this step must be repeated for all files resulting from the first demultiplexing step. Otherwise if an own version of Galaxy is used, it is possible to use another barcode splitter to perform the demultiplexing.
Enhanced Demultiplexing - Barcode Splitter Version 1.1
This tool is not available on the main Galaxy server. To use it one has to run a separate
version of galaxy and install this tool to this version. In the galaxy toolshed it
is called "fastx_barcode_splitter_enhanced".
This tool might be very helpful if the user wants to modify the demultiplexed FastQ files
further or demultiplex more than one time. It can be found at "NGS: QC and manipulation"
-> "FASTQ Trimmer". The only visible difference to the other barcode splitter is the
version number which is here 1.1.
The tool is used exactly like the Barcode Splitter 1.0.0. The difference is that every
result file is added to the galaxy history and can be further modified. Therefore it is
easy to consecutively perform multiple demultiplexing steps. The produced files can be
downloaded one by one.
To easily load the created files into BiQ Analyzer HiMod, it is helpful to create a spreadsheet containing the paths to all the files. Such a spreadsheet starts with one line for comments and description. If this line is empty, it will be ignored. The following lines need to be tab-delimited and contain a sample name, the path to a reference file and the paths to one or two read files, depending on the kind of analysis that should be done. Each possible combination of sample and amplicon needs to be listed in one independent line. In case there are no reads for a probe, the according line can end after the reference.
- Goecks, J, Nekrutenko, A, Taylor, J and The Galaxy Team. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010 Aug 25;11(8):R86.
- Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. "Galaxy: a web-based genome analysis tool for experimentalists". Current Protocols in Molecular Biology. 2010 Jan; Chapter 19:Unit 19.10.1-21.
- Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, Miller W, Kent WJ, Nekrutenko A. "Galaxy: a platform for interactive large-scale genome analysis." Genome Research. 2005 Oct; 15(10):1451-5.
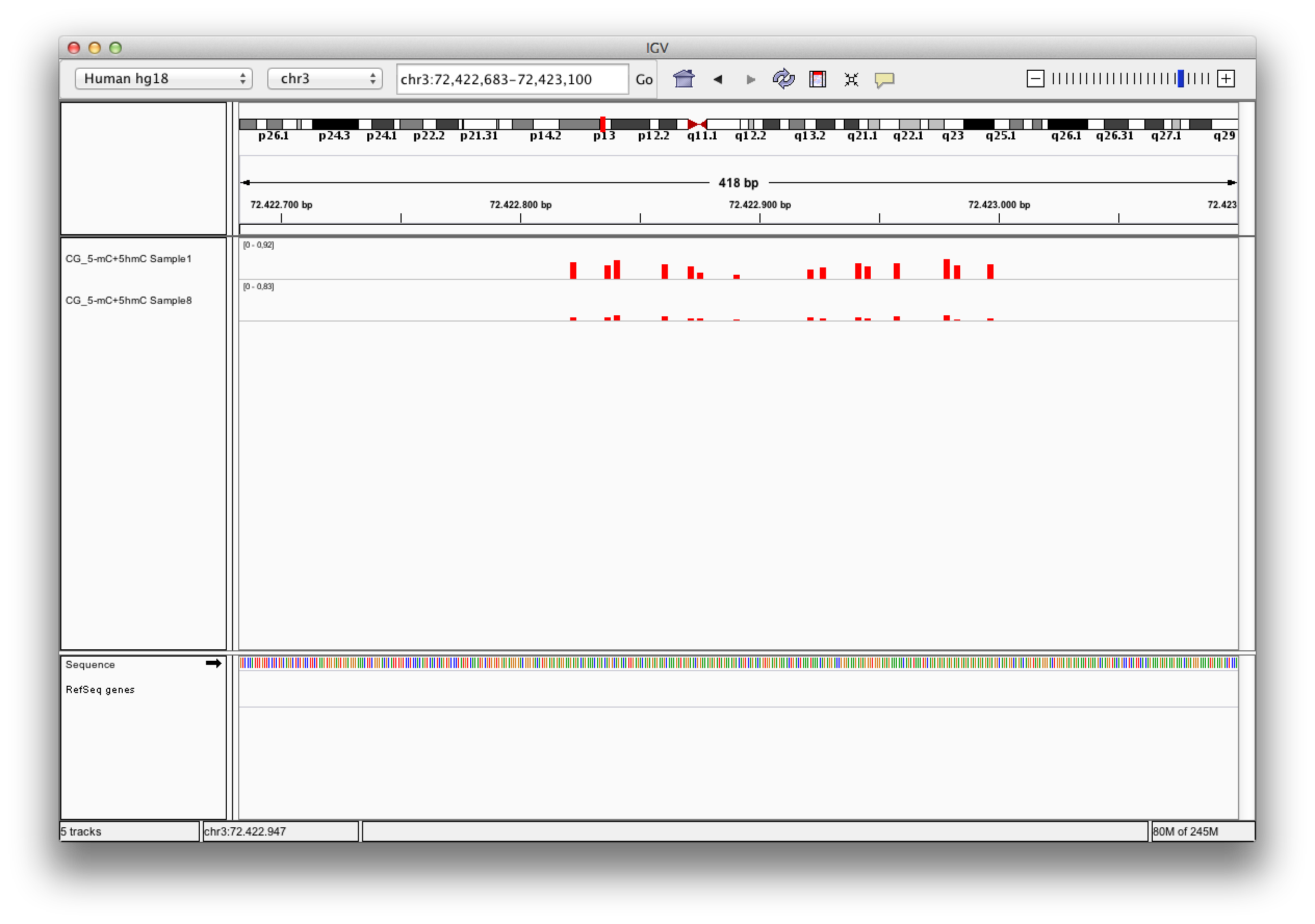
Q: What do I do with bedGraph files?
A: Exported bedGraph files can be used either with the IGV Browser or with the UCSC genome browser to visualize and compare modification levels with each other or other information given for that genetic region.
Each bedgraph file loaded into the IGV browser appears as a new track, containing information about the modification levels. Zooming in, one can review the data and compare different tracks at a position.
BiQ Analyzer FAQ
BiQ Analyzer doesn't work as expected? There are some hints on how to solve typical problems
Please check out all questions and answers below for a solution of your problem. In case you cannot find the answer, don't hesitate to write an e-mail to Christoph Bock. Your e-mail should include a detailed description of the problem, information on the operating system that you use and - if possible - some screenshots that illustrate the problem.
Please click on a question in order to expand the answer.
Installation Problems
Q: The installer doesn't work. What's wrong?
A: Check if you have really installed the latest version of Java. You have to have Java version 1.5 or newer installed on your computer, and it must be this version that is currently activated (in case you have more than one Java installation on your computer). Furthermore, on some computers you may have to start the installer from the command line, typing "java -jar install_BiQ_Analyzer.jar" while in the BiQ Analyzer directory.
Q: The program doesn't work at all on my machine. What can I do?
A: Carry out the installation procedure again and exactly as described - and be sure that you re-install the latest Java version, even if you have a recent version installed on your computer. If it still doesn't work, please write an e-mail to Christoph Bock with a detailed problem description.
Q: I want to install BiQ Analyzer on a Mac. What shall I do?
A: Be sure that you download the latest version of BiQ Analyzer because some older versions did not run properly on MacOS. Then follow the description on the "installation procedure" page.
Common User Problems
Q: The program does not recognise the CpGs in the genomic sequence. Why?
A: Are you sure that your genomic sequence has the right orientation (5' -> 3') and that you adjusted the "Conversion" choice box according to whether you are working on the plus strand (logical C to T conversion) or on the minus strand (logical G to A conversion)? Please check out the example in the Guided Tour!
Q: The program doesn't ignore my primers. That is not good, what can I do?
A: Unfortunately, the program doesn't know what your primers are (and even if it did, it would probably fail as soon as there are sequencing errors at the primers). Therefore it will be your duty to remove the primers manually. You can easily do so in the sequence text boxes on the left of the BiQ Analyzer window: just edit the sequences in the boxes and cut out the primers. These modifications will not be written back to the original sequence files.
Q: The multiple sequence alignment looks very messy: there is a line break
after the sequence name and each sequence occupies two lines. What shall I do?
A: This can happen if you have sequence names of unusual length. Just select
a smaller font size in the choicebox in the center of the BiQ Analyzer window
and everything should look alright again. Alternatively, there may be a problem
with font availability. In that case, please adjust the "Alignment font" entry in the configuration editor to a non-proportional font that is available on your system (e.g. "Courier New").
Q: For my sequences the "Reverse Complement" suggestions of the BiQ Analyzer are always wrong. Is the program stupid?
A: Yes and no, depending on your view. The program applies a simple sequence identity clustering in order to determine which sequences don't fit the genomic sequence. For the biggest cluster of those, it suggests to invert the sequences. This works very well when your sequences are sufficiently similar to the genomic sequence. But if you have strong deviations from the genomic sequence (e.g. due to many sequencing errors), this method is less successful. Unfortunately, the alternative (calculate another alignment with the inverted sequences and check whether this improves the fit) is time-consuming. Therefore, the best way to cope with this it to manually exclude highly erroneous sequences right after the first multiple sequence alignment and then press the "Recalculate" button to see if the situation improves.
Q: The multiple sequence alignment is very bad, even after reverse complement has been tried. What's wrong?
A: Maybe you selected G to A conversion instead of a C to T conversion or the other way around.
Q: When I copy the methylation information from the HTML output file and paste them into MS Excel, all information goes into the same cell. What shall I do?
A: This is because Excel tries to interpret the data as HTML instead of text. Use "Edit->Paste Special" and select "Text", then each field will go into a separate cell.
Q: What do all the colors in the multiple sequence alignment mean?
A: There is a file "Color Coding.pdf" in the BiQ Analyzer program directory that summarizes the color code.
Customization
Q: How can I change the minimum conversion rate and the minimum sequence identity that the program employs for quality control?
A: These values are defined in the configuration file of BiQ Analyzer and they can be changed using BiQ Analyzer's configuration editor. You can open that editor by clicking the "Config" button in BiQ Analyzer's main window.
Q: How can I change the default data path of the program?
A: Again, this value is defined in the configuration file of BiQ Analyzer and they can be changed using BiQ Analyzer's configuration editor.
Q: How can I change the experiment documentation questionnaire?
A: By default, the experiment documentation questionnaire template is defined by a file "ExpDetails_Template.txt" in the BiQ Analyzer program directory. This file can be changed using any text editor (but be sure to make a backup of the original file first - just in case you want to refer to it later on).
Data Security & Confidentiality
Q: The program connects to the internet on startup. Why does it do so? How can I disable it?
A: On each start, BiQ Analyzer contacts a server at the Max-Planck-Institute for Informatics to check whether there are new updates available. This is important because it will ensure that you receive the latest features and bug fixes when they become available (if the program finds a new update, it will politely ask you to install it, but never do anything behind your back!). The only piece of information that is transmitted to the update server is the version number (a string like this: "2.0"), no sequences, nothing else. However, if you want to disable this (for example if you don't regularly have an internet connection), you can do so in BiQ Analyzer's configuration editor: just tick the box for "Disable update check".
Q: What happens with my sequences when I choose to calculate the alignment remotely?
A: The raw sequences (and nothing else, especially not the experimental setting questionnaire) are sent to a ClustalW server at the Max-Planck-Institute for Informatics, where the multiple sequence alignment is calculated and passed back to the client. To do so, we have to save the sequences temporarily to our server - but they will not be used for anything apart from debugging errors in the program. If you are a company employee and your company policies prohibit to send any sequences over the internet please contact us and we will help you to set up a local BiQ Analyzer Remote Alignment Server.